
转录因子 (TFs) 是通过结合 DNA 来控制基因表达时空定位的蛋白质。在植物中,数十个 TF 家族与不同的结合位点 (TFBSs) 集合相互作用,这些位点反映了每个 TF 在生物体功能和物种特异性适应中的作用。然而,定义这些作用并理解调控演化的更广泛模式仍然具有挑战性,因为预测的 TFBS 可能对转录缺乏明确影响,且迄今为止实验获得的 TF 结合图谱规模不大或仅限于模式生物。在此,我们提出了一种可扩展的 TFBS 检测方法,并利用该方法创建了一个图谱集,涵盖了跨越 1.5 亿年开花植物演化的十个物种中 360 个 TF 的近 3000 个全基因组结合位点图谱。我们发现,远缘物种的 TF 直系同源物保持着几乎相同的结合偏好,而在相同的时间尺度上,TFBS 的获得和丢失却非常普遍。然而,在谱系内部,保守的 TFBS 占比过高,并存在于具有功能性调控元件特征的区域中。此外,具有保守 TFBS 的基因在 14 个单核 RNA 图谱中显示出细胞类型特异性表达的显著富集,为每个 TF 的活性和发育作用提供了可靠的标记。最后,我们比较了远缘谱系,阐释了古老的调控模块如何被招募和重连,以实现禾本科植物演化成功基础的适应性。

转录因子是识别并结合短且特定的DNA序列的蛋白质,它们调控基因在细胞中的表达时间、位置和方式。近期一项关于多种开花植物中转录因子结合的大规模研究表明,细胞类型特异的调控途径是保守的,而古老的调控网络为了谱系特异的表型(包括胁迫耐受性)发生了重新布线。
尽管植物在形态和功能上展现出巨大的多样性,但它们通常含有在序列水平上往往相似的核心基因集。已知转录因子(TF)活性有助于这些差异的形成,但特定调控程序内存在多少多样性一直不清楚。同样不清楚的是,转录因子本身的变异性与转录因子结合的顺式调控区域的变异性相比,对表型差异的相对贡献是多少。尽管这些问题在之前的小规模研究中已被探讨过,但它们在大规模研究中的影响尚未得到能够得出一般结论的阐释。在本期中,Baumgart等人承担起了探究转录因子结合活性如何随进化时间变化这一艰巨任务。
研究方法
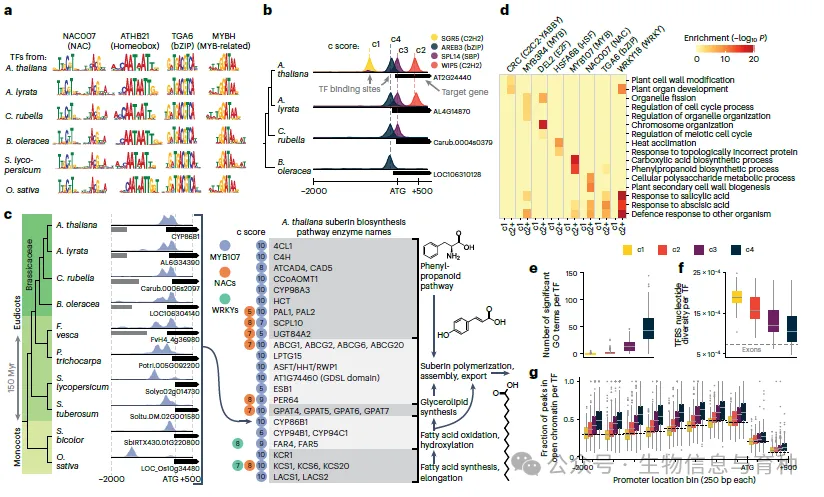
作者首先通过改编他们之前开发的multiDAP方法,该方法使用混合DNA来检测全基因组范围内的转录因子结合,以适应大小从125兆碱基(Mb)到约800Mb不等的大型植物基因组,最终生成了包含十个不同物种(包括单子叶植物和双子叶植物)的3000个转录因子结合数据集(图1)。这些数据集随后被用于比较同源转录因子在其同源基因组中的结合与拟南芥(Arabidopsis thaliana)转录因子的结合。作者发现,拟南芥转录因子的结合事件与使用来自不同物种的同源转录因子与其自身DNA测试得到的结合事件高度相似。这使他们得出结论,即使来自同一结构家族的高度不同的转录因子,其同源转录因子的结合在很大程度上也是保守的。
在确定了拟南芥转录因子可以作为远缘植物物种的代表性转录因子后,他们接下来测试了结合靶标的保守性和变异性,首先在十字花科(Brassicaceae)家族内的近缘亲属中进行,然后是比较更远的物种。在近缘亲属的实验中,使用了代表33个不同家族的244个拟南芥转录因子,然后使用multiDAP方法和从四种不同的芸苔属植物(包括拟南芥)中提取的基因组DNA进行测试。将来自不同物种的目标基因(如果峰值位于起始密码子上游2000个碱基对(bp)以内至下游500个bp以内,则视为目标基因)分配到“直系同源组”,并根据特定转录因子在各种基因组中结合直系同源基因的次数分配一个“保守分数”(c分数)。仅在单个物种中结合的转录因子-目标基因对被赋予c1的保守分数,而在四个芸苔属基因组中结合的则被赋予c4的保守分数。这种分类使作者能够得出结论,在110万个拟南芥转录因子-直系同源组相互作用中,24%在所有四个芸苔属物种中高度保守(c4),尽管非编码调控序列高度不同,以至于无法进行序列比对。这与21%的拟南芥转录因子-目标基因对被评为私有靶标(c1)形成了对比。这表明,虽然转录因子结合位点(TFBS)随时间的推移已经分化(即私有位点可能已经丢失或获得),但在近缘物种中,转录因子-目标基因关系有很大比例是保守的。有趣的是,保守的转录因子目标基因显示出更强的功能基因本体富集,更可能与可访问的染色质区域重叠,并显示出核苷酸多样性降低的迹象,这表明在c4位点存在纯化选择。这些发现表明,可以使用保守分数与转录因子结合来划定功能调控区域并鉴定重要的转录因子-目标基因关系。
接下来,使用较小的转录因子子集,该子集跨越了包括草莓、杨树、马铃薯、番茄、高粱和水稻以及四个芸苔属基因组在内的十个远缘基因组,将这一框架应用于更远的物种。在更长的时间尺度上,观察到了类似的趋势,即7.1%的芸苔属c4转录因子-目标基因关系在所有10个物种中保守(c10分数)。这些“超保守”的转录因子-目标基因组包括控制蜡质生物聚合物(对形成各种类型植物细胞壁中的保护屏障至关重要)生产的核心途径、细胞分裂相关过程、根毛发育、胁迫和干旱反应。综上所述,这些实验优雅地表明,尽管转录因子结合位点比转录因子结合特异性的分化程度要高得多,但一套核心的转录因子-目标基因集在至少1.5亿年的时间尺度上被保留了下来。
进一步分析
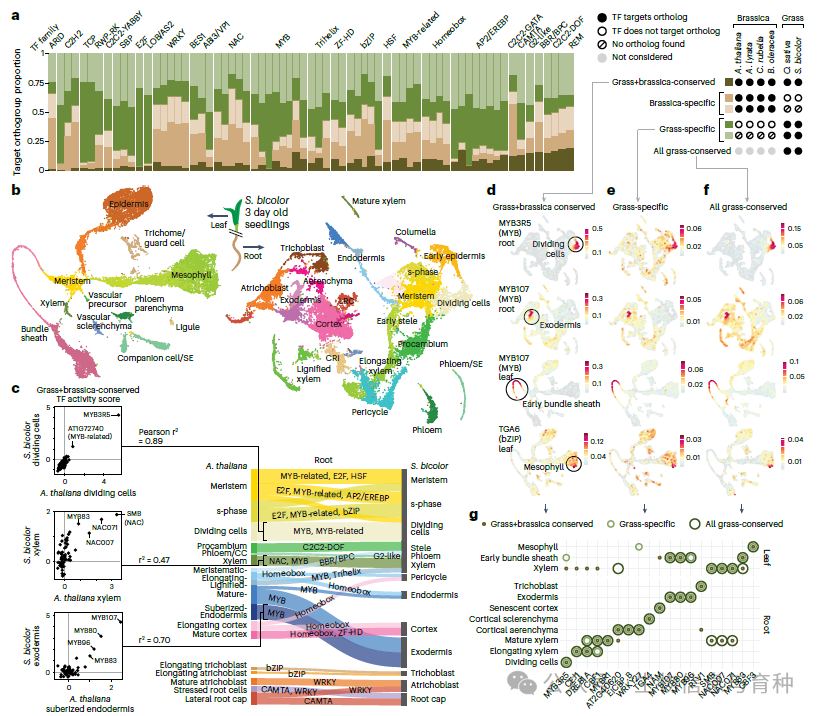
在确定了共有的谱系特异或保守的转录因子目标基因集后,作者通过对每个芸苔属物种的各种组织进行单核RNA测序(snRNA-seq)来剖析它们的表达谱。以拟南芥基因组的数据作为初始基线,他们确定单个转录因子及其目标基因显著共表达,且最保守的目标基因(c4)中的显著性水平最高。有趣的是,这一分析还显示,c4目标基因比c1、c2或c3目标基因更具细胞类型特异性,并且在几种情况下,这一趋势也适用于超保守的转录因子-目标对,目标基因在高度不同的物种中维持了相同的细胞类型特异性表达。此外,作者还能够使用这种方法将转录因子的调控活性映射到所有四个物种的特定细胞类型中,从而可能鉴定出细胞类型特异性表达的重要调控因子。
使用基于c分数的方法,作者还比较了四个芸苔属谱系的转录因子结合位点与两个草谱系(高粱和水稻)的转录因子结合位点。他们鉴定出所有六个物种共有的核心调控子(由同一转录因子调控的一组目标基因),其中许多显示出强烈的细胞类型特异性表达。此外,为了探索功能的保守性或分化,他们产生了高粱茎和根的单核RNA测序数据集。这使他们能够鉴定出草谱系和芸苔属谱系特有的转录因子-目标对,并提供了转录因子网络广泛重新布线的证据,包括在高粱中将参与蜡质生产的靶标募集到叶鞘细胞中,在那里它们有助于高粱的干旱耐受性。综上所述,这些结果支持了一个模型,即某些核心调控子在1.5亿年的进化过程中持续存在以控制各种细胞类型特异的过程,而其他顺式调控区域似乎已被重新布线以适应谱系特异的功能。

研究意义与展望
尽管这项研究专注于基因组大小小于900Mb的植物物种,但许多其他具有经济重要性的作物物种(例如玉米、小麦、大麦和棉花)拥有更大的基因组,其调控元件通常位于距目标基因更远的距离。将Baumgart等人使用的方法扩展到这些和其他物种,并探索基因组扩张如何影响转录因子活性的保守性和重新布线以形成重要的农艺性状,将是非常有趣的。对multiDAP策略进行额外的修改,以纳入额外的调控区域中的DNA甲基化状态或多聚体转录因子复合物,可能进一步加深我们对转录因子调控网络进化复杂性的理解。总体而言,这项杰出的研究为不同开花植物中转录因子调控网络在单细胞水平上的进化提供了急需的见解。这些知识对于理性地改造作物物种的性状以及为未来的生物技术策略提供信息至关重要。